GATK: A much more sophisticated option for variant calling
...
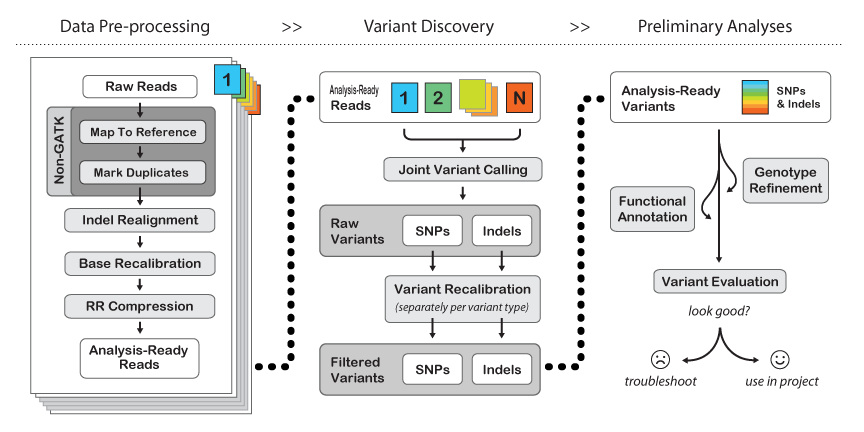
Here is their general workflow:
(from: http://www.broadinstitute.org/gatk/guide/best-practices)
Pipeline explanation:
- The first few steps are "Non-GATK" - meaning they are just what we have already been doing in the course. You may "map to reference" using either BWA or bowtie2, but you will find BWA used more often with GATK. BE SURE TO MAP WITH READ GROUPS.
- The Mark Duplicates step serves the purpose of pre-identifying PCR duplicates in your data so that the genotyper doesn't get skewed by potential amplification artifacts when weighing support for a variant. If your library is reasonably complex and not vastly over-sampled, this should only knock out about 10-20% of your raw data. If it takes out a higher percentage of your data but you still have significant coverage (e.g. >30x at minimum, or >50x on average) then you should still be fine. If you have many samples, you can get away with lower per-sample coverage since "Variant Discovery" is done jointly across the entire sample set.
- Indel realignment - turns out that insertions & deletions can lead to artifacts like SNPs being called at their edges just because the mapper can't quite figure out where the in/del should be relative to a SNP on a per-read basis. There are two steps to this - "RealignerTargetCreator" that figures out where realignment should be done and then "IndelRealigner" that does the re-alignment. It's a two-step process because you can use external information (e.g. the indel catalog provided by the 1000 genomes project) to more sensitively detect possible indel sites.
- BaseRecalibrator - re-scores the per-base quality scores to be more accurate. Raw quality scores in your fastq files come from aggregate measures of images, sequencing-by-synthesis intensities, spatial variation on the flow cell, etc. Re-calibration takes the mapped raw data, checks how good it is against the given reference AND a list of common SNPs (like dbSNP for human), builds a new error model with this "training set" and provides a "recalibration table" that subsequent GATK tools can use to tweak base quality values.
- RR compression is optional - just saves disk space by intelligently throwing away data that is not required downstream. We have plenty of space at TACC, so I recommend not using it.
- Joint Variant Calling can be done by one of two tools: HaplotypeCaller or UnifiedGenotyper. HaplotyperCaller is the go-to tool for human genomes & exomes. UnifiedGenotyper is more tweak-able - better for tumor studies (e.g. mixed populations) and if you may have ploidy > 2. Here is a great blog entry by Craig DeLoughery comparing the two.
- Variant Recalibration is the same song, different verse, to Base Recalibration.
- GATK has an extensive set of tools for filtering and functional annotation. SPHS likes many other ways of doing this better (like grep/awk/sed/sort/join for filtering, and annovar for annotation), but if you're sticking to human only analysis then the GATK tools and suggested workflows are probably a good idea.
...
The best option is to use the ones in $BI if they are there (in $BI/ref_database/GATK) (NOTE: You can join the BioITeam and put them there!! Free storage this way!). Otherwise make your own local copy from those provided by GATK. TACC does not maintain these for the community (yet).
Expand title Tips on downloading, setting up GATK bundles... To pull GATK resource bundles and check them:
ftp ftp.broadinstitute.org
username: gsapubftp-anonymous<find bundle you want>
prompt
bin
mget *
Then: cd to dir, fix md5 files:
for file in `ls *.md5`; do sed -i 's/\(\/\).*\(\/\)//' $file; done
do md5 check:
for file in `ls *.md5`; do md5sum -c $file; done
- If you are in a non-model system, you are on your own to make the resource bundle. For instance, if you have a fasta file of your reference then you'll need to use the picard tool CreateSequenceDictionary.jar to make a dictionary of it for GATK. Use "module load picard" and then "ls $TACC_PICARD_DIR" to see all the picard tools at TACC.
...