...
- mode union. This mode is recommended for most use cases.
- mode intersection-strict.
- mode intersection-nonempty.
 Image Removed
Image Removed
...
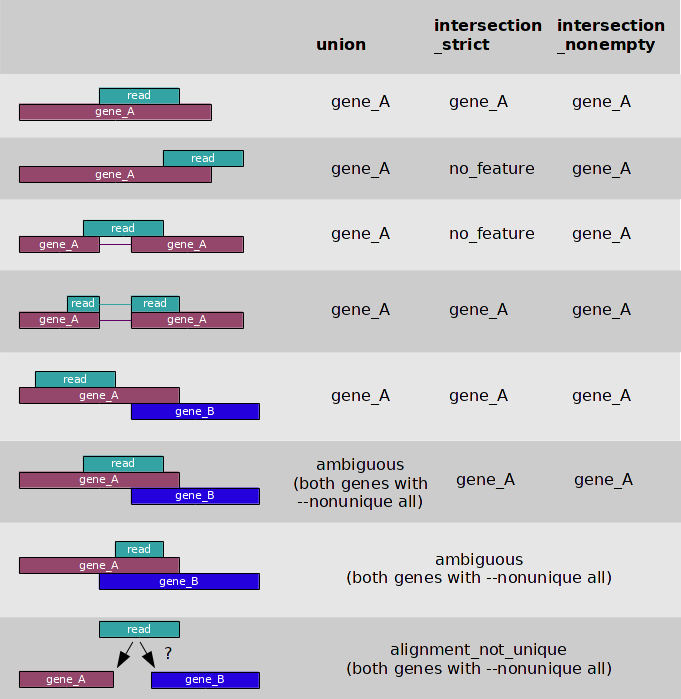 Image Added Image from:http://www-huber.embl.de/users/anders/HTSeq/
Image Added Image from:http://www-huber.embl.de/users/anders/HTSeq/
What happens to the amiguous reads depends on this flag:
--nonunique none (default): not counted for any features.
--nonuniqueall: counted towards all.
Installing HTseq
Htseq is NOT a module on lonestar6. Module spider htseq doesn't find anything, so we have to install it.
| Code Block |
|---|
|
module spider htseq
|
...
Generally, installing tools to a cluster is a pain, so avoid it if you can. However, if you have to install something, remember that you cannot install things on TACC globally, so you'll have to use options to install the tool locally to a directory that is accessible to you. Detour to how to install tools locally
...
| Warning |
|---|
| title | Warning: To submit to queue |
|---|
|
| Code Block |
|---|
nano commands.htseq
#put these lines in the commands file
htseq-count -m intersection-nonempty --stranded reverse -i gene_id hisat_results/C1_R1.sam ../reference/genes.formatted.gtf > C1_count1.gff
htseq-count -m intersection-nonempty --stranded reverse -i gene_id hisat_results/C1_R2.sam ../reference/genes.formatted.gtf > C1_count2.gff
htseq-count -m intersection-nonempty --stranded reverse -i gene_id hisat_results/C1_R3.sam ../reference/genes.formatted.gtf > C1_count3.gff
htseq-count -m intersection-nonempty --stranded reverse -i gene_id hisat_results/C2_R1.sam ../reference/genes.formatted.gtf > C2_count4.gff
htseq-count -m intersection-nonempty --stranded reverse -i gene_id hisat_results/C2_R2.sam ../reference/genes.formatted.gtf > C2_count5.gff
htseq-count -m intersection-nonempty --stranded reverse -i gene_id hisat_results/C2_R3.sam ../reference/genes.formatted.gtf > C2_count6.gff |
| Code Block |
|---|
launcher_creator.py -j commands.htseq -n htseq -q normal -t 02:00:00 -a OTH21164 -l htseq_launcher.slurm -m "module load htseq"
sbatch --reservation=RNAseq htseq_launcher.slurm |
AFTER THIS COMPLETES: | Code Block |
|---|
join C1_count1.gff C1_count2.gff| join - C1_count3.gff | join - C2_count4.gff |join - C2_count5.gff|join - C2_count6.gff > gene_counts_HTseq.gff
#if you have many samples, use for-loop and join |
|
...