| Table of Contents |
|---|
...
Here is their general workflow:
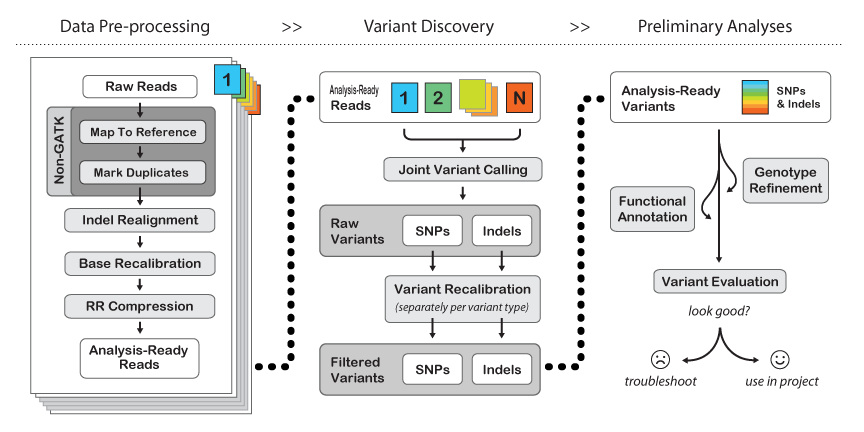
(from: http://www.broadinstitute.org/gatk/guide/best-practices)
Objectives:
- Provide a fairly detailed understanding of a common human genome variant analysis program.
- Provide a working analysis pipeline and sample data to evaluate said pipeline.
...
The first few steps are "Non-GATK" - meaning they are just what we have already been doing in the course. You may "map to reference" using either BWA or bowtie2, but you will find BWA used more often with GATK. BE SURE TO MAP WITH READ GROUPS.
The Mark Duplicates step serves the purpose of pre-identifying PCR duplicates in your data so that the genotyper doesn't get skewed by potential amplification artifacts when weighing support for a variant. If your library is reasonably complex and not vastly over-sampled, this should only knock out about 10-20% of your raw data. If it takes out a higher percentage of your data but you still have significant coverage (e.g. >30x at minimum, or >50x on average) then you should still be fine. If you have many samples, you can get away with lower per-sample coverage since "Variant Discovery" is done jointly across the entire sample set.
Indel realignment - turns out that insertions & deletions can lead to artifacts like SNPs being called at their edges just because the mapper can't quite figure out where the in/del should be relative to a SNP on a per-read basis. There are two steps to this - "RealignerTargetCreator" that figures out where realignment should be done and then "IndelRealigner" that does the re-alignment. It's a two-step process because you can use external information (e.g. the indel catalog provided by the 1000 genomes project) to more sensitively detect possible indel sites.
BaseRecalibrator - re-scores the per-base quality scores to be more accurate. Raw quality scores in your fastq files come from aggregate measures of images, sequencing-by-synthesis intensities, spatial variation on the flow cell, etc. Re-calibration takes the mapped raw data, checks how good it is against the given reference AND a list of common SNPs (like dbSNP for human), builds a new error model with this "training set" and provides a "recalibration table" that subsequent GATK tools can use to tweak base quality values.
RR compression is optional - just saves disk space by intelligently throwing away data that is not required downstream. We have plenty of space at TACC, so I recommend not using it.
Joint Variant Calling can be done by one of two tools: HaplotypeCaller or UnifiedGenotyper. HaplotyperCaller is the go-to tool for human genomes & exomes. UnifiedGenotyper is more tweak-able - better for tumor studies (e.g. mixed populations) and if you may have ploidy > 2. Here is a great blog entry by Craig DeLoughery comparing the two.
Variant Recalibration is the same song, different verse, to Base Recalibration.
GATK has an extensive set of tools for filtering and functional annotation. SPHS likes many other ways of doing this better (like grep/awk/sed/sort/join for filtering, and annovar for annotation), but if you're sticking to human only analysis then the GATK tools and suggested workflows are probably a good idea.
...
- The documentation appears extensive, but it's written for experts - you have to know a lot to make use of their documentation.
- Load
There is only one command, "GenomeAnalysisTK.jar" which as you can see is a Java (64-bit) Jar file, so to run anything the syntax is:
Code Block java -Xmx4g -jar $TACC_GATK_DIR/GenomeAnalysisTK.jar -T <GATK module you want to run> <other options>
Implications of this, and other notes:
- Watch out for Java memory requirements: -Xmx4g has always worked for me. Someday it might not.
- Tools usually have "inherited options". These are options that are used across many tools. They are not documented on the tools page but on the GATK command line page. This is a neat idea, but makes it hard to track down details for a specific command.
- Worse than having split documentation, options aren't always consistent. For instance, for the IndelRealigner tool uses "-known" while the RealignerTargetCreator uses "--known" (two dashes) instead even though you run them back-to-back.
- Multithreading options are really tricky - they may or may not be supported on any given tool.
- Broad is constantly working on the software. Things like the names of options, parameter values, etc. may change over time (though it is increasingly stable) so watch out if you upgrade.
- Unlike most programs which fail very quickly (i.e. seconds) if you give them bad input, GATK takes a while to fail - it can take a minute or two just to get a tool going to the point that it fails, and the error will be buried in a lot of other verbose output.
- Pay attention to parameters and be willing to invest significant time tuning these to your specific application. The GATK documentation specifically says to do this - don't use it blindly (like any complex tool).
- Almost all the tools are deathly slow - pretty much any step done in GATK will take a long time. For instance, when I run human exomes at 40 million 2x100 read-pairs per sample, mapping takes only a few hours but the rest of the GATK pipeline can take another 24 hours on a single sample.
- There are two ways to "run" GATK tools: a) command line mode, one tool at a time, and b) pipeline mode. - which assumes you are running the LSF job scheduling system, consequently it's not used at TACC (Lonestar uses SGE and Stampede uses Slurm).
...
The best option is to use the ones in $BI if they are there (in $BI/ref_database/GATK) (NOTE: You can join the BioITeam and put them there!! Free storage this way!). Otherwise make your own local copy from those provided by GATK. TACC does not maintain these for the community (yet).
Expand title Tips on downloading, setting up GATK bundles... These are very advanced commands and listed here only as a reference. To pull GATK resource bundles and check them:
username: gsapubftp-anonymous<find bundle you want>
prompt
bin
mget *
Then: cd to dir, fix md5 files:
for file in `ls *.md5`; do sed -i 's/\(\/\).*\(\/\)//' $file; done
do md5 check:
for file in `ls *.md5`; do md5sum -c $file; done
- If you are in a non-model system, you are on your own to make the resource bundle. For instance, if you have a fasta file of your reference then you'll need to use the picard tool CreateSequenceDictionary.jar to make a dictionary of it for GATK. Use "module load picard" and then "ls $TACC_PICARD_DIR" to see all the picard tools at TACC.
...
| Expand | |||||
|---|---|---|---|---|---|
| |||||
|
A working (on Lonestar) GATK pipeline script
| Expand | |||||
|---|---|---|---|---|---|
| |||||
This short script will run all the many GATK commands on a SINGLE bam file - that's not best practices if you have more than one sample, but it's easy to modify this to do the first few steps separately, then join your BAMs (samtools merge) and run the last few steps. WARNING: GATK is slow - running this script on only 12 million reads (6 million read-pairs) takes about 2 hours on Lonestar!
|
Compare samtools to GATK on exome 20.
Diversions
These are oddities:
...